Hematologia en 3 minutos: Aprendamos de Sickle Cell Disease
Brandow, A.M., Liem, R.I. Advances in the diagnosis and treatment of sickle cell disease. J Hematol Oncol15, 20 (2022)
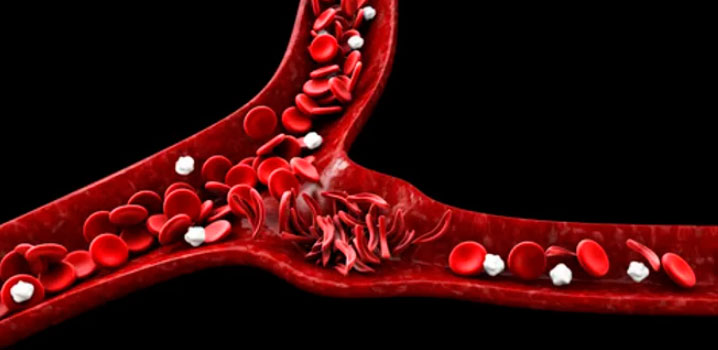
La anemia de células falciformes (ACF) es una hemoglobinopatía (hemoglobina enferma) que se produce por una mutación que afecta el gen que codifica la cadena b de la hemoglobina (b-hb) y afecta a cerca de 3 millones de personas en el mundo.
La enfermedad asociada (Sickle cell disease o SCD) se caracteriza por anemia hemolítica crónica coombs negativa, dolor agudo y crónico así como daño de diferentes órganos que afectan la calidad y sobrevida de los afectados con mediana de mortalidad cercana a los 43 años.
Podría interesarte
Hematologia en 3 minutos: Aprendamos de Sickle Cell Disease
Brandow, A.M., Liem, R.I. Advances in the diagnosis and treatment of sickle cell disease. J Hematol Oncol15, 20 (2022)
La anemia de cé...
iBTK en Linfoma del Manto. Esperanzas de sobrevida con menor toxicidad.
El linfoma del manto (LCM) es una neoplasia linfoproliferativaB que puede tener un comportamiento agresivo, requiriendo en muchas oportunida...

Leucemia Linfática Crónica: Rol de Acalabrutinib en 1° linea
Acalabrutinib con o sin antiCD20 tiene mejores resultados que obinutuzumab+clorambucil en pacientes mayores de 65 años ± comorbilidades (s...